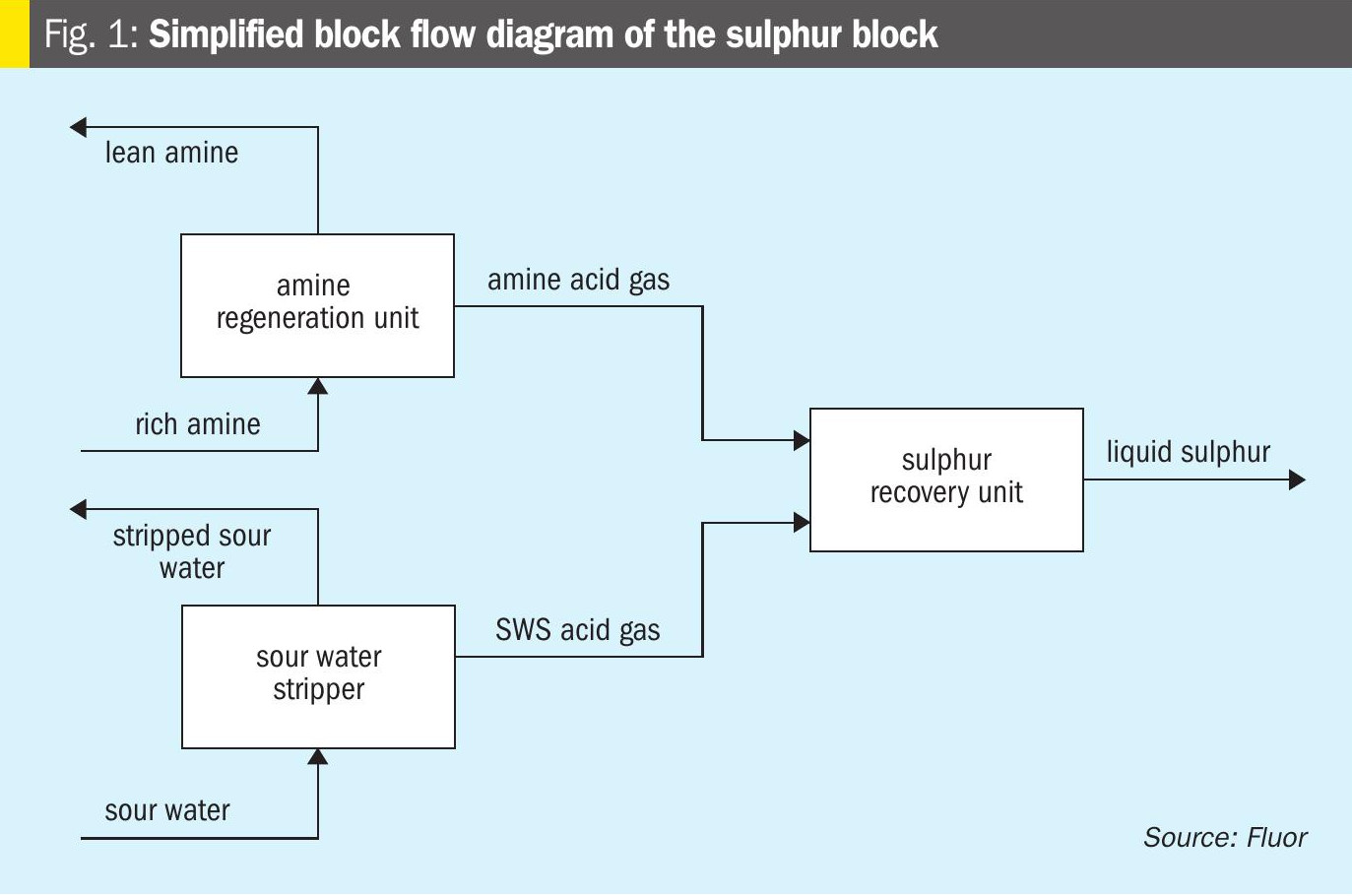
Sulphur 421 Nov-Dec 2025

24 November 2025
Neutralisation of heat stable salts – Part 1
AMINE SYSTEMS
Neutralisation of heat stable salts – Part 1
Joel Cantrell of Bryan Research & Engineering and Clay Jones of INEOS GAS/SPEC re-evaluate the historical practice of intentionally adding strong base such as caustic (NaOH) to amine with the purpose of improving performance and reducing corrosion by “neutralising” heat stable amine salts (HSAS) which have accumulated in the amine.
Neutralisation with caustic in amine systems has long been a debated topic in the gas sweetening industry. Neutralisation is the practice of intentionally adding a strong base (NaOH, Na2CO3 , KOH, etc) to amine for the purpose of improving performance and reducing corrosion by “neutralising” heat stable amine salts (HSAS). While this article is not likely to resolve all differing opinions on this topic, the authors hope that it will serve as a useful reference by summarising the current state of understanding.
The article will approach the topic in four sections which are supported by simulation case studies and plant data as applicable:
- Chemistry review to explain theoretical considerations and list relevant chemical reactions which will be referenced throughout the rest of the paper.
- Heat stable salt (HSS) effects on amine chemistry and corrosion.
- NaOH effects on amine chemistry and corrosion.
- Effects of adding too much NaOH.
The article has been split into two parts, Part 1 features the first two sections, with the remaining two sections in Part 2, which will be published in the next issue of Sulphur.
For brevity’s sake, this article focuses on a representative subset of the chemicals which are currently in use in industry. The only acid gas addressed extensively is H2S, the only amine considered is MDEA, and the only strong base discussed is NaOH. While there are other acid gases, amines, and strong bases that are important in industry, it is hoped that all of the relevant points can be made more effectively by focusing on the underlying chemical principles rather than juggling a larger list of similar reactants. The main conclusions of this paper are:
- HSS are common contaminants in amine units. They increase corrosion rate, chemically bind or neutralise amine, and often they accumulate over time. Dealing with HSS is a typical challenge in operating an amine unit.
- There are several established methods for handling HSS. Neutralisation is one option. It has the goal of neutralising HSAS (converting HSAS into Inorganic HSS) and extending the time before the more effective (and expensive) methods are used. Neutralising does not get rid of HSS anions, but tries to mitigate some of their negative effects.
- Published studies and reports from operating plants are not unanimous about whether or not neutralising reduces corrosion rate. However, there are several effects which are generally accepted to happen:
- Neutralising can convert bound amine to free amine, which restores incremental acid gas pickup capacity.
- Neutralising can increase lean loading. In some cases, this can hurt the solvent’s treating performance.
- Injecting too much or too fast creates a risk of precipitation, plug-gage, and alkaline stress corrosion cracking (SCC).
- Neutralising is not a permanent solution for ongoing HSS contamination. It is intended to be a coping mechanism for extending the time between reclaim/purge events. Neutralisation is commonly used as one part of a comprehensive HSS management strategy.
- A significant number of operating sites neutralise their amine, and contend that they get benefits which outweigh the risks.
Chemistry review
The conclusions of this paper rest on several concepts from chemistry, which are reviewed here for convenience.
Electrolyte chemistry concepts
There are several important electrolyte, or ionic, chemical reactions which happen in the aqueous amine solutions used in gas treating. These reactions are responsible for the primary function of the amine: absorb acid gas in the contactor then release the acid gas in the regenerator.
For example, equation 1 and equation 2 below demonstrate how H2S is chemically trapped in an MDEA solution. In equation 1, aqueous H2S lives up to the name “acid gas” by giving an H+ ion to the solution. This renders the remaining HS– ion as completely non-volatile and chemically traps the H2S in solution. In a complementary reaction, MDEA acts as a base by accepting H+ ions to create a protonated MDEA molecule, MDEAH+ .
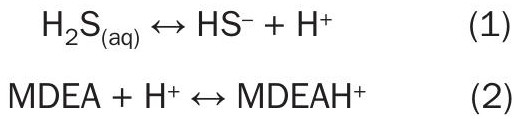
These reactions underscore two important concepts:
Firstly, equation 1 is the underlying mechanism for chemically capturing acid gas in amine. Ionic charged species such as HS– are not volatile; they are chemically trapped in solution. Conversely, non-charged molecules are volatile to the extent allowed by vapour pressure or Henry’s constant. This is a key concept for how acid gases get captured in amine, but in the context of this article, it also drives the influence of neutralisation on the volatility of HSS species in the regenerator’s reboiler. This effect is also responsible for one of the undesired consequences of over-neutralisation, which is elevated lean loadings.
Secondly, reactions affect each other through the common ion effect. In water, equation 1 does not proceed very far to the right. However, the presence of MDEA greatly increases the amount of H2S which can be held in the aqueous phase by a factor on the order of 1000x. This is accomplished through Le Chatelier’s principle: If an equilibrium is disturbed, the system will shift to counteract that change and establish a new equilibrium. In the case of equation 1, as H+ ions are produced by H2S, those ions are captured by MDEA through equation 2. This drives equation 1 further to the right, making it harder and harder for the volatile H2S(aq) molecules to exist in solution, since they’re preferentially converted to non-volatile HS– ions after giving up their H+ ions to MDEA. Le Chatelier’s principle will be important in several aspects of the discussions below.
Ionic vs covalent bonds
The following discussion draws heavily on Chapter 3 of Reference 1.
The term chemical bond encompasses two distinct modes through which atoms are held together: covalent bonding and ionic bonding.
The term covalent bond refers to the bonding mechanism in which the outer orbitals of the bonded atoms overlap, resulting in increased electron density between the atoms. There are several sub-types of covalent bonds, including the metal-ligand bonds discussed later in this paper. The term ligand refers to molecules which bond to a central metal atom. All atoms are not created equally when it comes to covalent bonding: some atoms are much more available for covalent bonding due to their electronic configuration, whereas others are less able to do so. For example, the Fe2+ cation can readily form coordinate covalent bonds with various ligands when it is in an aqueous solution, because it is a transition metal and therefore has d-orbitals in its outer shell. In contrast, the Na+ cation’s outer electronic configuration is much less available for covalent bonds.
The term ionic bond refers to bonds where atoms of opposite charge are attracted to each other by electrostatic forces. The concept of an ionic bond is an idealisation which does not perfectly reflect real world solutions. In actuality, all ionic bonds also include some amount of covalent bonding, though the relative importance of those covalent bonds is much smaller.
Cation behaviour in solution
Two cations are of special interest for the purposes of this article: iron and sodium. These species behave quite differently in amine solutions. The Fe2+ cation enters the solution through corrosion, and Na+ might be intentionally added during neutralisation, or it may unintentionally enter the amine through contaminated makeup water, leaking isolation valves, amine reclamation etc.
When Fe2+ enters the solution, it enters as a complex surrounded by water molecules. Because of its electronic configuration, which includes the d-orbitals of a transition metal, each central Fe2+ cation is surrounded by six water molecules. The water molecules serve as ligands, as shown below in equation 6 and equation 8. The six ligands organise into an octahedral complex around the cation. Each ligand shares a coordinate covalent bond with the central iron atom. Both electrons of the bond are donated by the ligand. The metal-ligand bonds are true chemical bonds with measurable bond enthalpy. The solubility of a metal is strongly influenced by the stability of the metal-ligand bonds. As will be discussed, ligands can be substituted: the water molecules can be replaced by other molecules such as HSS anions.
In contrast, the electronic configuration of Na+ cations is relatively inert with respect to covalent bonds. In fact, the outer shell of Na+ cation has the same configuration as the noble gas neon. Given its electronic configuration, Na+ is not very likely to have orbital overlap or participate in covalent bonds. However, it has a concentrated electrostatic charge in a relatively small volume, so it exerts considerable influence on polar and charged molecules in its vicinity. Approximately 16 to 17 water molecules are attracted to each Na+ cation to such an extent that they can be considered as bound to the Na+ with an ionic / electrostatic force.
That being said, coordinate covalent bonds with Na+ cations still form at a detectable concentration, though they are less favoured than bonds with transition metals such as Fe2+. For example, reference 3 documents the critical stability constant for complexes of K+ cations with oxalate and complexes of Na+ cations with acetate. By analogy, complexes of Na+ and oxalate should be expected to exist as well.
Corrosion reactions
Iron corrosion proceeds through the anodic, or oxidation, reaction listed in equation 3. Typically, in amine systems, the corresponding cathodic, or reduction, reaction involves protonic acids, which provide H+ ions that react as shown in equation 4. Adding these two reactions together, we get equation 5 which shows a simple corrosion reaction for iron. The H+ ions in equation 5 can come from the surrounding solution or from protonic acids like H2S or HS– .
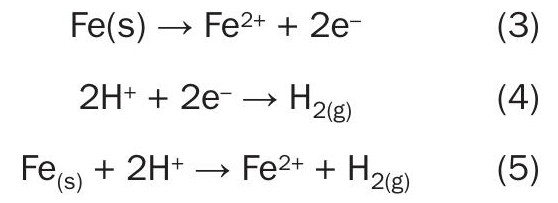
Iron cations from equation 5 can go into solution surrounded water ligands as shown in equation 6. In an amine system with H2S present, it is also likely that the iron could be converted into a solid corrosion product as in equation 7, with HS– serving as the source of H+ for corrosion. (Recall that chemically captured H2S primarily exists as HS– in amine solutions.) Iron sulphide created in equation 7 also has some solubility, which can be expressed as equation 82.

Equation 6 shows the general principle that everything else being equal, more acidic solutions will be directionally more corrosive to iron. Equation 7 shows how amine solutions with higher acid gas loading become more corrosive, and also how captured H2S leads to the formation of solid iron sulphide. Iron sulphide can be deposited as a layer coating the inner surface of pipes and equipment; it can also (through equation 8) form as a particulate floating in the amine. The adhered layer of solid iron sulphide is known to significantly slow down the overall corrosion rate by slowing down mass transfer between the bulk amine solution and fresh, uncorroded iron underneath.
Equation 8 is a reversible reaction. This fact is one reason for the common observation that even when the lean amine is clear when it enters the absorber, the rich amine leaving the absorber will often have a significant amount of iron sulphide particles leading to dark or green appearance. In the absorber, as the amine picks up H2S, the equilibrium of equation 8 will naturally shift to the left due to increasing concentration of HS– (aq) ions, causing more of the dissolved iron cations to precipitate as FeS solids.
Ligand substitution and chelation
As discussed above, in amine solutions, dissolved iron cations exist as the central metal atom surrounded by ligands, which are covalently bonded to it. For many metal atoms, these ligands will be water as shown in equation 6 and equation 8. However, other species can displace water to serve as a ligand. Equation 9 shows an example of a ligand substitution reaction where formate (HCOO– ) enters the complex to become a ligand. While equation 9 shows one water molecule being displaced, it is also possible for more than one displacement to happen for the same central iron cation.
Researchers have catalogued equilibrium constants for reactions such as equation 9, which can be found in published tables of critical stability constants, for example Reference 3. Substitution reactions such as these are one way that HSS anions increase the net corrosion rate: they stabilise iron cations in solution, i.e., they shift equation 6 and equation 8 further to the right.
An additional effect, called chelation, happens when one ligand molecule is able to form more than one bond with a central metal cation. The oxalate anion, which is sometimes present as a contaminant in amine solutions, is an example of a chelating anion. Each oxalate anion (C2O42-) has two carboxylic acid groups, each of which carries a -1 charge. Oxalate can displace two water molecule ligands as shown in equation 10. Ligands in chelate structures make more stable complexes. This stability is at least partially due to statistical/entropic considerations: If a random encounter leads to a bond with one of oxalate’s sites, then the oxalate’s second bonding site will stay in close proximity and have a much better chance of creating a second bond. The reverse reaction, i.e., displacement of one oxalate ion by two water molecules, is similarly less likely because it requires the concurrent displacement of two bonds. The chelation effect explains why, relative to other HSS anions, oxalate has a significantly higher critical stability constant with iron3 and has been observed to lead to significantly faster corrosion rate in amine solutions, as shown in Fig. 1.
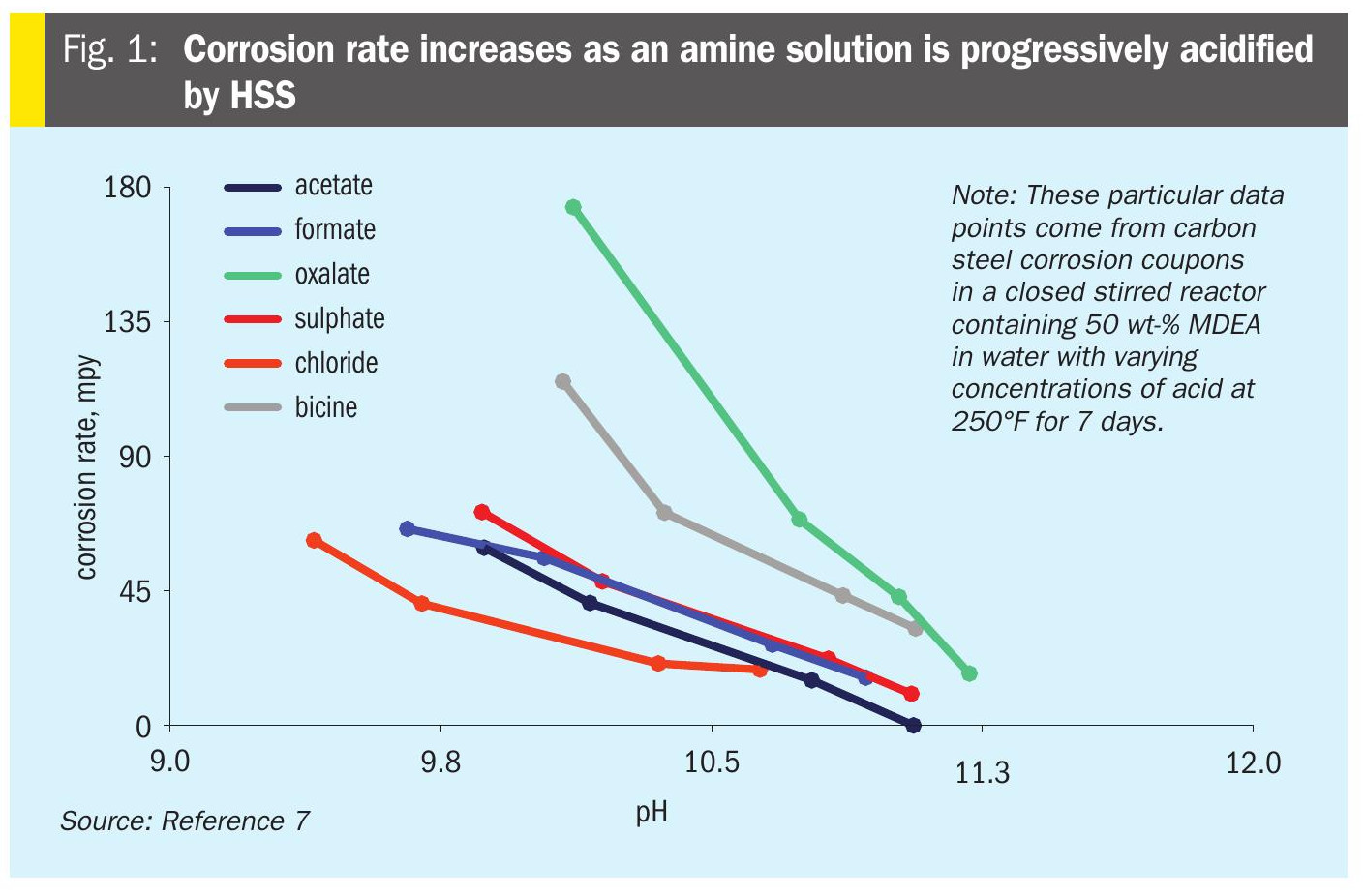
Soluble metal vs corrosion rate
Even though soluble metal concentrations are frequently reported for amine solvents, there is some nuance to understanding what this measurement tells us. There is not a rigorous direct relationship between the concentration of metal held in solution and the corrosion rate experienced in an amine unit. The corrosion rate in an amine unit depends on many parameters which are well documented in literature, for example in Reference 4. In general, the corrosion rate will vary quite significantly from one place to another within the same amine unit. Local factors typically govern the severity and location of the worst corrosion in the unit. For example, the following factors are common culprits in corrosion failures: high rich loading, high velocity/turbulence from flashing 2-phase flow, high wall shear due to mechanical design that creates eddies and/or impinging flow5. These local factors must be identified through a unit review and cannot be identified through laboratory analysis of the amine.
The effect of the various local corrosion factors are additive in an operating unit. A unit which has a step change increase in rich loading will have an incremental increase in corrosion rate in the rich amine circuit. Similarly, a unit which has a step change increase in soluble iron concentration should be expected to have incrementally faster corrosion rate. The significance of any specific increase in corrosion rate will include considerations of the remaining corrosion allowance in the specific pieces of equipment, the quality of the inspection program in the plant, etc.
The cycle of taking Fe2+ cations from the internal surface of equipment/pipe, then precipitating that iron as FeS fouling is described as an “iron pump” in Reference 8. Under this paradigm, we can think of a solvent that holds more iron as a higher capacity iron pump. Therefore, we should expect a directional relationship between iron solubility and corrosion rate: higher soluble iron concentration is directionally worse than lower soluble iron concentration. Nonetheless, it is not easy to determine when the difference is significant enough to justify taking action. Good unit monitoring practice is to watch trends of soluble metals over time and to interpret step changes or shifts as potentially serious signs of increased risk.
Important chemical reactions
The list of chemical reactions below shows the primary reactions in a typical amine solution. Acetic acid, H3C2OOH, is included as a typical HSS. These reactions are listed here as an introduction and for easy reference in the rest of the article.
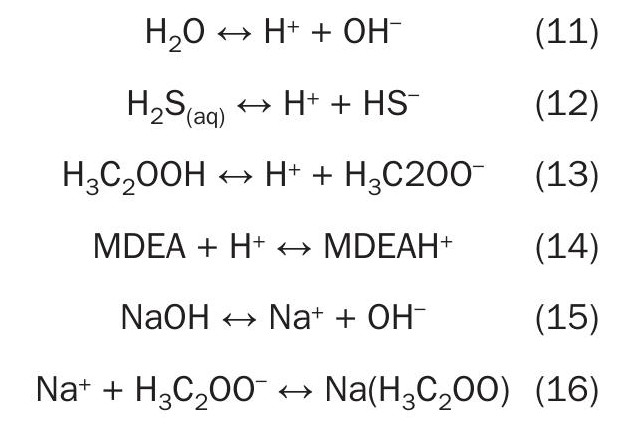 One important feature of this reaction set is that the acid H2S and stronger acids such as acetic acid H3C2OOH, do not react directly with the amine MDEA. Instead, these acids donate H+ cations to the solution, and the basic amine MDEA accepts H+ cations from the solution. It is important to realise that the H+ cations in solution are all equivalent to each other regardless of which molecule they came from. This relationship between different reactions is called the common ion effect; it is the mechanism through which the concentration of one ionic species affects the concentration of another across a network of reactions.
One important feature of this reaction set is that the acid H2S and stronger acids such as acetic acid H3C2OOH, do not react directly with the amine MDEA. Instead, these acids donate H+ cations to the solution, and the basic amine MDEA accepts H+ cations from the solution. It is important to realise that the H+ cations in solution are all equivalent to each other regardless of which molecule they came from. This relationship between different reactions is called the common ion effect; it is the mechanism through which the concentration of one ionic species affects the concentration of another across a network of reactions.
For example, consider the impact of increasing the amount of acetic acid H3C2OOH in a solution with H2S and MDEA. Acetic acid is a stronger acid than H2S. Increasing the concentration of acetic acid in solution will increase the concentration of H+ ions, which in turn will both increase the extent of MDEA’s protonation (equation 14 moves further to the right) and it will also decrease the extent of H2S dissociation (equation 12 moves further to the left). Both of these effects of HSS (i.e., more protonation of amine, less dissociation of acid gas) will be discussed further soon.
Another key aspect of this set of reactions is that NaOH can be assumed to completely dissociate into Na+ and OH– in the aqueous solution. These dissociated ions are not tightly bound to each other, so they will move independently through the solution. The reader is encouraged to look through the list of reactions and identify the ones which involve Na+ and OH–. In the case of OH–, its only reaction is the reversible reaction with H+ to make water. (OH– also appears in equation 7 and equation 8, but these are less significant side reactions due to the low concentration of soluble iron, which is typically 10s of ppmw Fe or less.)
Reactions between Na+ and HSS anions, such as equation 16 are not extensively documented, but they do seem to happen. These complexes are important for considering corrosion phenomena related to neutralisation. Any HSS anions which are complexed with Na+ ions will be less available for complexing with iron in reactions such as Equation 9 and equation 10. Therefore equation 16 is a mechanism for neutralisation to slow down corrosion.
Evidence for reactions such as equation 16 can be found in the literature. Reference 3 gives the critical stability constant for a complex of potassium cation with oxalate anion; presumably complexes of sodium and oxalate also exist, though they are not listed. Reference 9, which focuses on bicine corrosion, documents that critical stability constants for complexes between alkali metals and amino acids are available from the public data resource NIST SRD 46 – which the authors of the present paper were not able to confirm due to time and IT constraints (the freely available NIST SRD 46 database is accessed through an executable compiled to run on Windows XP or earlier). The authors of Reference 9 note that the NIST database does not contain a critical stability constant for complexes of sodium and bicine, but it does contain constants for complexes of sodium with related or analogous amino acids. They reasonably conclude that, by analogy, some amount of coordinate covalent bonding would be expected between sodium and bicine as well.
HSS effects on amine chemistry and corrosion
More acidity, more iron complex stability
It is well-documented in industry that amine solutions become more corrosive as HSS anions accumulate. This general principle can be seen in Fig. 1 which shows how corrosion rate increases as an amine solution is progressively acidified by HSS. This lab study did not reflect all aspects of typical industrial conditions – especially the presence of H2S. Since there was no H2S present to create a protective iron sulphide film in the experiments, an important factor that influences corrosion rate in real world amine units was missing. Nonetheless, there is a clear relationship for each acid showing that corrosion rate increases as pH falls. In the limiting case of a completely absent or destroyed FeS passivation layer, this corrosion behaviour could be expected. This study also shows that oxalate and bicine are significantly more corrosive than other anions at the same pH, which is attributed to the ability of oxalate and bicine to chelate iron.
Other studies2,6 present data showing an analogous relationship between increasing HSS concentration and increasing corrosion rate in the presence of H2S and CO2. Since the accumulation of HSS will reduce the solution’s pH (more H+ available, less OH– available), corrosion rate from equation 6 and equation 8 should naturally increase. Additionally, HSS will increase corrosion rate by stabilising iron in solution through reactions similar to equation 9 and equation 10. This effect will act to stabilise any iron cations in solution, regardless of how they got there, but it is thought to be especially damaging to process equipment in amine service because – through the common ion effect and equation 8 – increased HSS concentrations damage the protective iron sulphide layer which has a strong influence on overall corrosion rate.
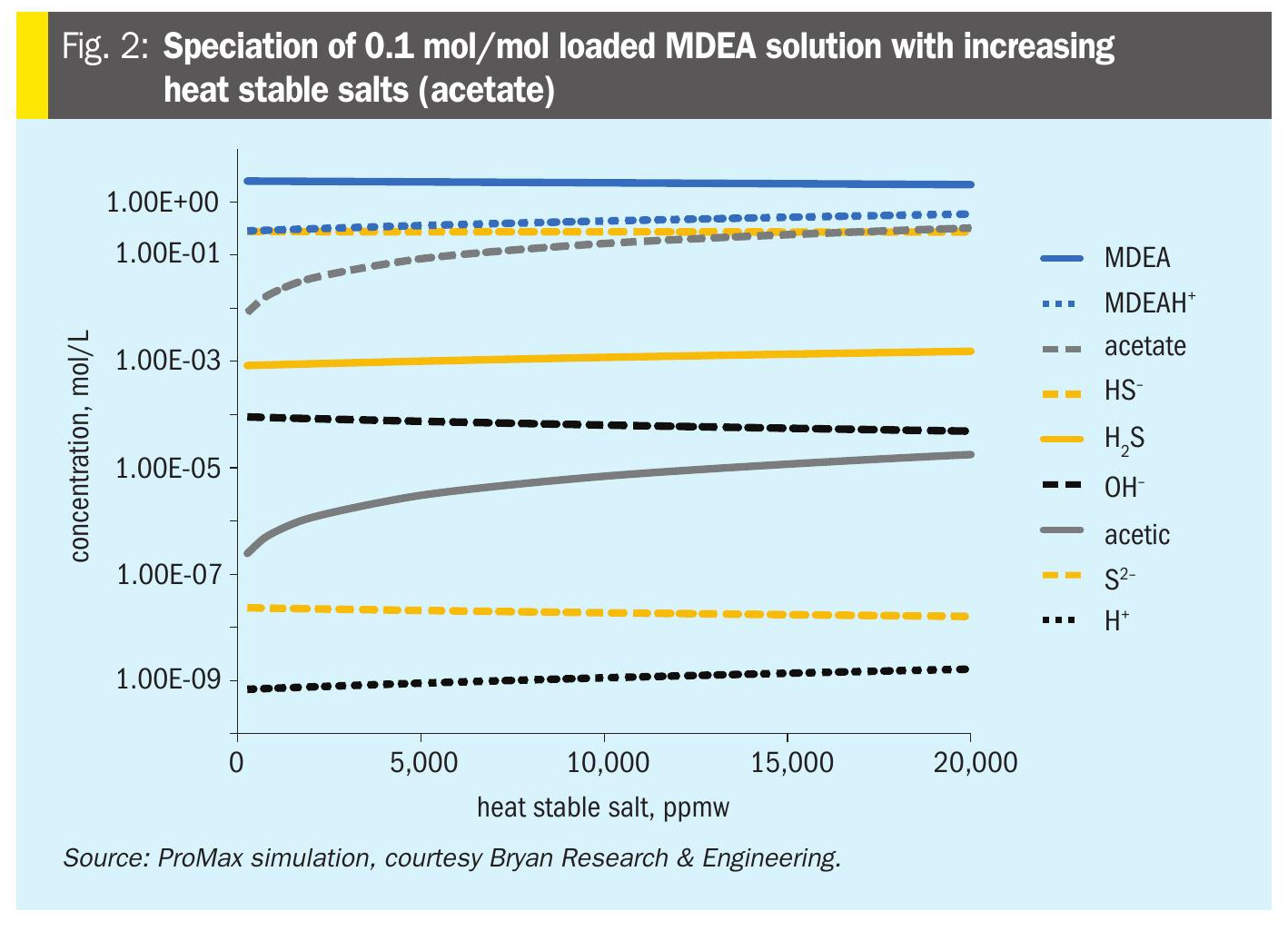
Fig. 2 shows the effect of HSS on the speciation of a rich MDEA solution with increasing acetate content, based on ProMax simulation. The acetate (and acetic acid) increase, causing a rise in H+ and a decrease in OH–. Increasing H+ increases MDEAH+ at the expense of MDEA. The HS– remains roughly constant, while H2S rises and S2– falls. These results demonstrate how increasing amounts of HSS lead to more bound amine (more MDEAH+) and less solubility of H2S (molecular uncharged – and therefore volatile – H2S is a larger fraction of total H2S).
Volatile acid in reboiler
There is another mechanism for HSS-driven corrosion, which specifically affects the bottom of the regenerator, the reboiler, and the reboiler vapour return line. This mechanism involves increased volatility of some acids as the overall concentration of HSS increases. Equation 12 and equation 13 show how acids will find an equilibrium between the volatile molecular form and the non-volatile ionic form. The fraction of molecules that exists in volatile vs non-volatile forms is dictated by the temperature-dependent pKa value for each acid. Of course, the reason that we call these acidic contaminants Heat Stable Salts is that under normal operating conditions they are almost completely trapped in the non-volatile ionic form and cannot be steam stripped from the amine to a significant extent. However, some of the weaker acids (such as acetate with pKa = 4.76 and formate with pKa = 3.75) can sometimes achieve small but significant volatility in the regenerator’s reboiler. Volatile acids pose a particular corrosion risk in the reboiler because the acid vapour can be absorbed into droplets of water which condense on the inner surface of the reboiler shell, reboiler vapour return line, etc. Since these water droplets contain much less amine than the bulk circulating solvent, the pH of the droplets is much lower than the bulk solvent, which can lead to increased corrosion rate. This effect is explored further later. This effect will be explored further in Part 2 of this article.
Beneficial side effect: Lower lean loading
Increasing concentration of HSS contaminants results in increased acidity (less basicity) of the amine solvent. As discussed above, increased acidity naturally shifts the ionic equilibrium of captured acid gas towards its volatile molecular form, with the net effect of making it easier to strip acid gas out of the solvent and achieve a lower lean loading. This effect is a natural consequence of equation 12. In some units (especially TGUs), this effect can improve unit performance, but in other units the effect will not provide a significant benefit.
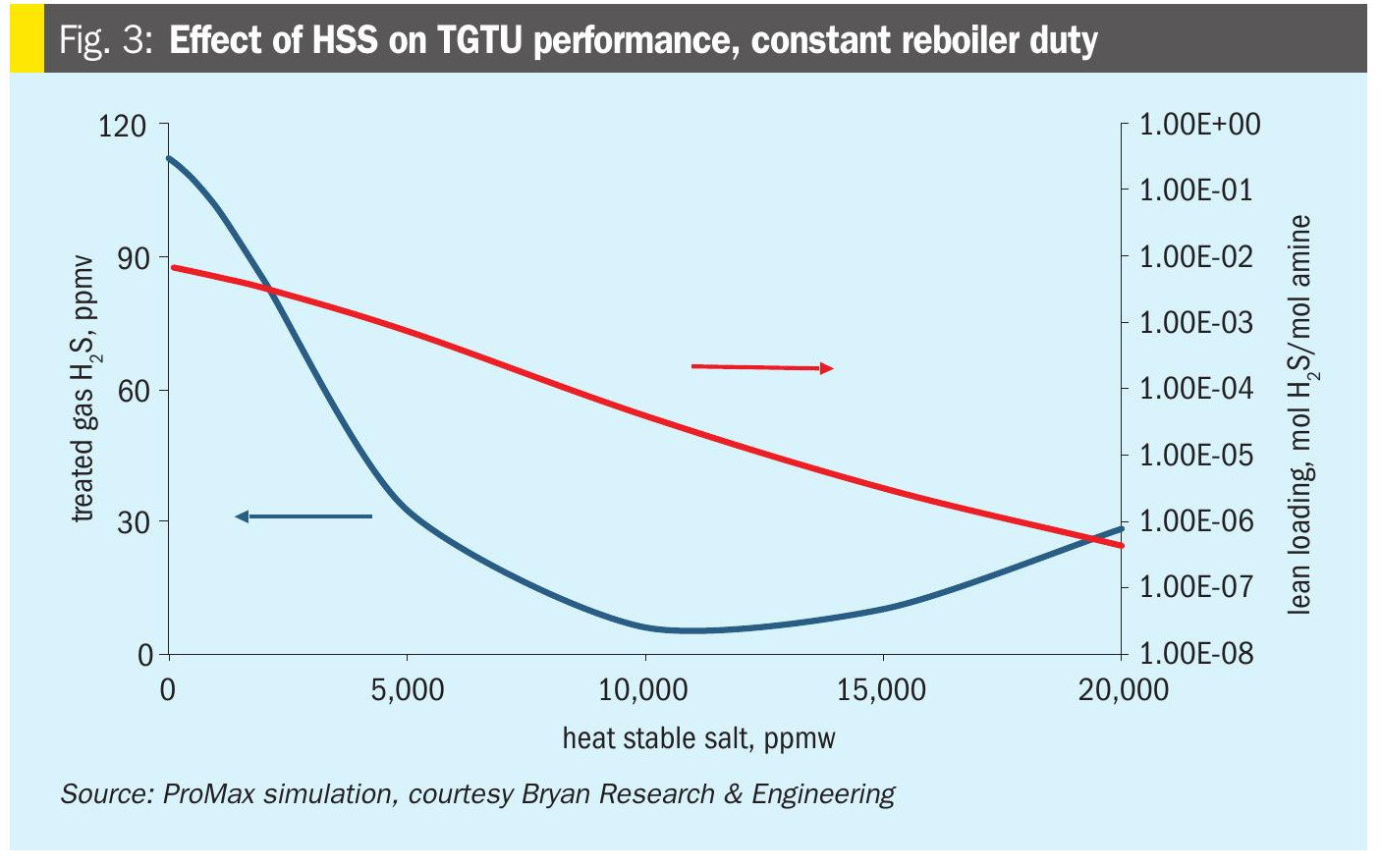
Fig. 3 shows the simulated impact of increased HSS on the overall behaviour of a typical TGU. For a constant reboiler duty, as HSS increases, the lean loading goes down. This lower lean loading improves treating performance to a point. After sufficient MDEA is tied up in HSAS, the treating performance is degraded and the treated H2S content increases.
References
This article will be continued in the January-February 2026 issue of Sulphur magazine.